Researcher Profiles

Christine R. Zhang, Ph.D.
2023 Funding recipient
Targeting DNA damage repair to harness DNMT3A-mutant clonal hematopoiesis and MDS development
EvansMDS Young Investigator Award 2023
PROJECT SUMMARY
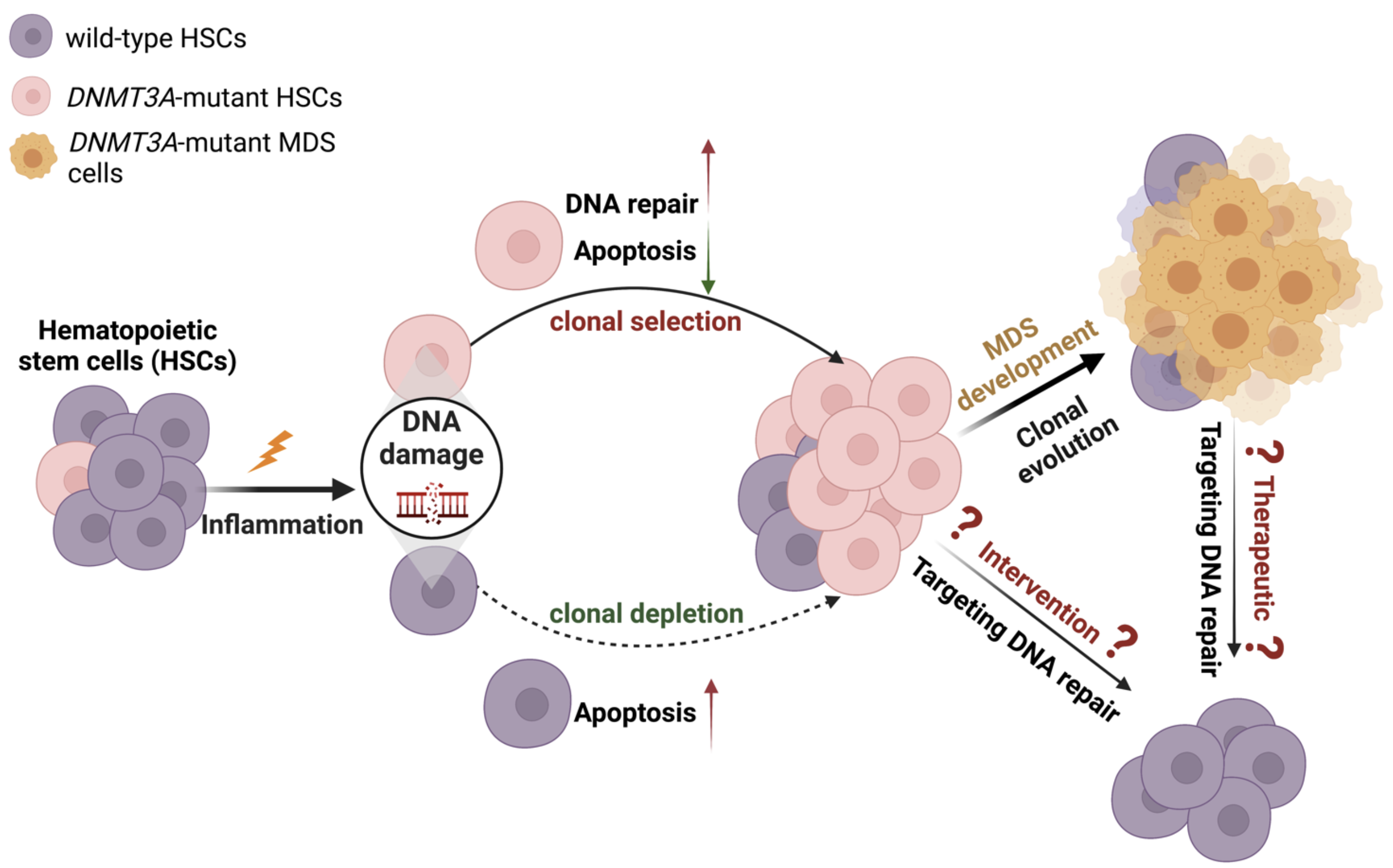
Our goal is to understand how DNA mutations cause myelodysplastic syndromes (MDS) and provide preventive and therapeutic opportunities for the patients. MDS is a group of common blood malignancies that are often associated with an enhanced inflammatory condition, and are caused by ineffective blood production due to somatic mutations acquired in the hematopoietic stem cells (HSCs). Many MDS mutations are populated in the DNA Methyltransferase 3A (DNMT3A) gene, causing protein functional loss. Approximately 10% of MDS patients harbor DNMT3A mutations that often predict a poor prognosis, a higher risk of leukemic transformation, and an overall shorter survival. While there is an urgency for better therapies for DNMT3A-mutated MDS patients, the pressing need has been impeded by an incomplete understanding of how the loss of functional DNMT3A gives rise to MDS development. The proposed research here will better understand the role of DNMT3A functional loss in the MDS development and how MDS development and progression can be prevented.
Cumulative evidence has suggested that inflammatory stress may prime DNMT3A-mutant clonal hematopoiesis into MDS development. We previously reported that chronic type II interferon signaling selects for DNMT3A– mutant HSCs while causing wild-type HSCs exhaustion. This fitness advantage of the mutant HSCs was attributable to the resistance to HSC activation and apoptosis. Interestingly, activated mutant HSCs remain apoptotic-resistant during interferon signaling stress. These data suggest additional mechanisms protect the proliferative mutant HSCs from interferon signaling associated depletion. Our preliminary data have indicated that proliferative mutant HSCs may repair inflammation-induced DNA damages more efficiently, at least partly attributable to the increased non-homologous end-joining DNA repair pathway activity. Inhibiting this pathway sensitized the mutant but not control mouse MDS-like blasts to interferon signaling induced cell death. Therefore, we hypothesize that targeting this DNA repair pathway may abolish the selective advantage of DNMT3A mutant HSCs during inflammatory stress and thus minimize the risk of MDS development and progression. Herein, we propose directly assessing the therapeutic potential of targeting DNA repair (NHEJ) in the clonal expansion and MDS development mouse models, as well as primary patient samples. We will also explore the molecular dependencies of the increased NHEJ in the mutant HSCs during inflammatory stress to identify possible targets for specific elimination of DNMT3A-mutated HSCs as an early intervention/therapeutic strategy to minimize MDS risks/treat DNMT3A-mutated MDS patients.