Researcher Profiles

Sushree S. Sahoo, Ph.D.
2023 Funding recipient
Elucidation of molecular mechanisms in SAMD9L-related pediatric marrow failure and MDS
EvansMDS Young Investigator Award 2023
PROJECT SUMMARY
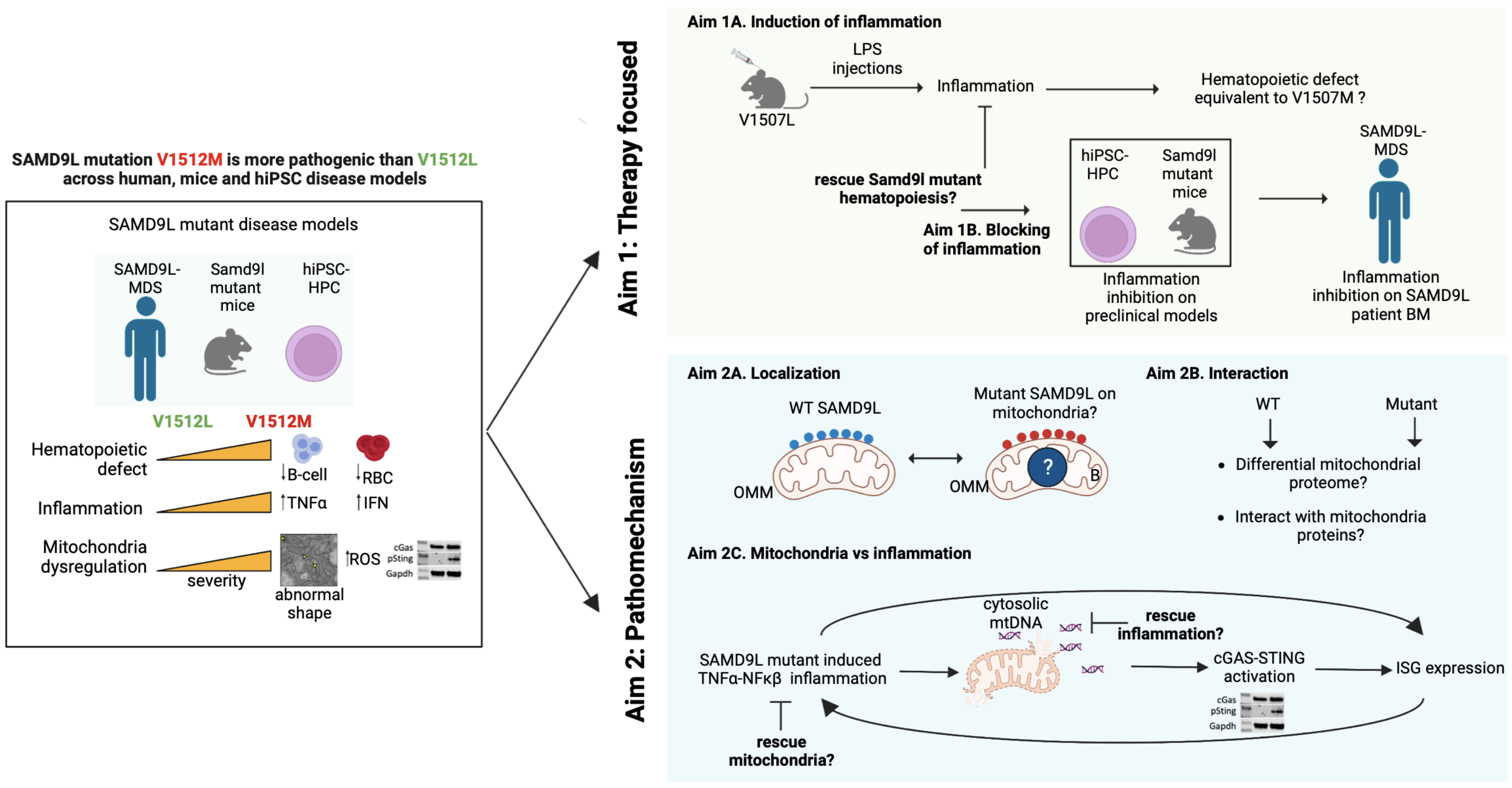
Childhood myelodysplastic syndrome (MDS) is a rare disorder where the blood-making cells don’t work properly. Germline mutations in two interferon-responsive genes, SAMD9 and SAMD9L (SAMD9/9L), are responsible for causing MDS in 8-20% of all cases. We know that the mutant proteins stop cells from growing significantly higher than the wildtype, which can lead to protein synthesis arrest and cell death. But we don’t know the underlying mechanism or why the mutant protein can cause an immune response that leads to a bad outcome. Right now, bone marrow transplant is the only way to help the sickest children. To find better treatments, we need to do more research to determine how SAMD9/9L mutant proteins are causing MDS. To do this, we need to make models with mutations that adequately mirror the wide clinical spectrum of the disease. Toward this, I have mapped the genetic landscape of SAMD9/9L mutations. I have identified a recurrently mutated SAMD9L amino acid, Valine 1512, which manifests a severe or mild clinical phenotype in humans when changed to Methionine or Leucine. Using human-inducible pluripotent stem cells and mice models with endogenous V1512M or V1512L mutations, I show SAMD9L mutation-specific TNFa and IFN inflammatory signatures and mitochondria stress, observed with increased severity in V1512M mutation compared to V1512L. In the first part of the study, I propose a comprehensive set of studies to assess if the variability in the blood phenotypes of mutant SAMD9L result from different degrees of inflammation and, if inhibited, can rescue the blood production defect. By inducing more inflammatory stress in the mild V1507L mice (ortholog of V1512L), I aim to assess if it recapitulated the severe blood and stem cell phenotypes of V1507M mice (ortholog of V1512M). Using a battery of pharmacologic and genetic inhibitors on hiPSC, mice, and patients’ bone marrow cells, I will examine if specific inhibition of inflammation can normalize the SAMD9L mutant-induced blood defect. These experiments will rigorously test the feasibility of inflammatory blockade as a therapeutic strategy for SAMD9L-associated MDS. Next, I will investigate how SAMD9L mutations cause inflammation. Based on my findings of WT SAMD9L localizing to mitochondria and the association of SAMD9L mutations with abnormal mitochondrial biology, I will investigate the pathophysiologic role of mutant SAMD9L in mitochondria and how it can contribute to MDS in patients. Utilizing my established, biochemical and cellular fractionation assays, I will interrogate the physiological link of SAMD9L mutant proteins with mitochondria. I will investigate mutant protein localization and interaction with mitochondria proteome. Finally, I will tie my inflammation and mitochondria stress observations to a proposed working model of SAMD9L-associated MDS disease pathogenesis where TNFa causes mitochondrial injury, leading to the release of mitochondria DNA (mtDNA) to the cytosol and cGAS-STING activation and expression of interferon-stimulated genes (ISG). Thus, forming a feedforward loop of inflammation and mitochondria injury. I will investigate this working model by depleting mtDNA and assess if inflammation is alleviated, and reciprocally, I will inhibit TNFa and assess if mitochondria phenotype is improved. Our approach to studying MDS predisposition due to inherited SAMD9L mutations holds the unique potential to uncover key insights into the complex mechanisms of how these gain-of-function mutations affect hematopoiesis and cause MDS predisposition.