Researcher Profiles

Gang Huang, Ph.D.
Cincinnati Children's Hospital Medical Center
2020 Funding recipient
A novel strategy targeting metabolic reprogramming to intervene in transformation from MDS to secondary AML
Discovery Research Grant 2020
PROJECT SUMMARY
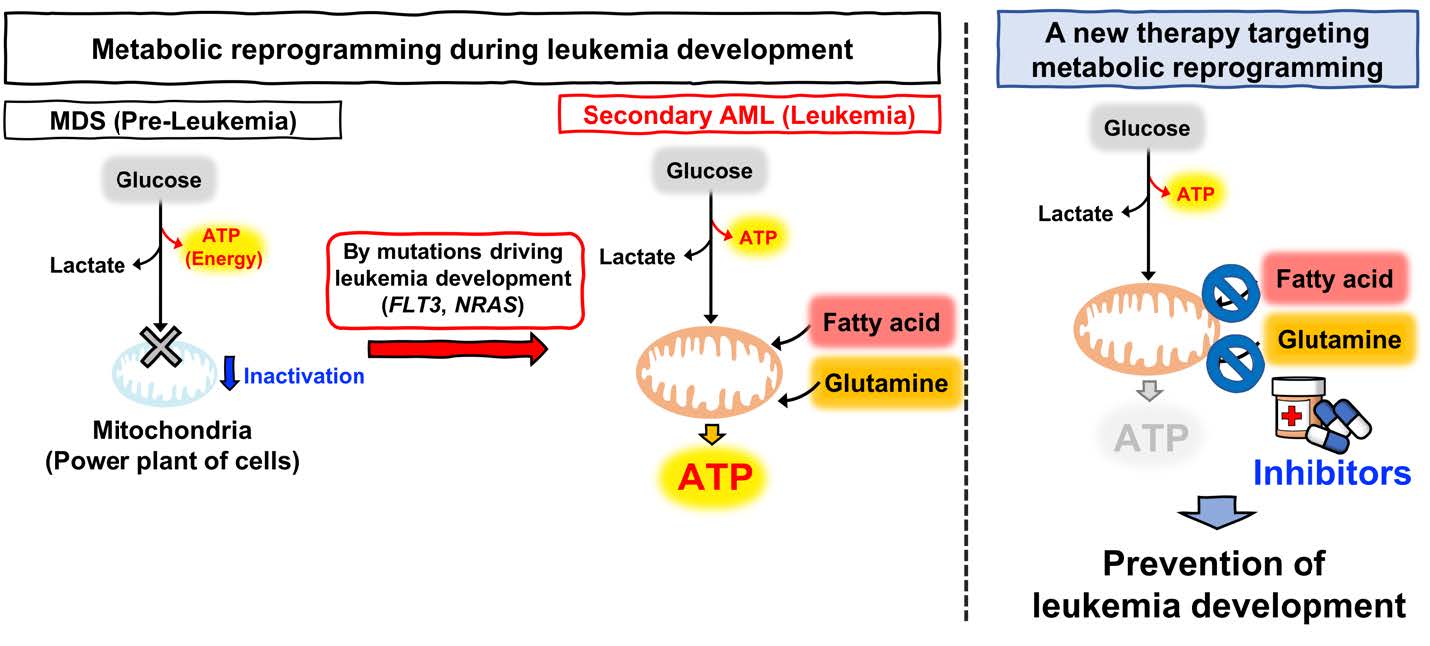
Secondary acute myeloid leukemia (sAML) is developed during the disease progression of myelodysplastic syndromes (MDS) clones and is associated with acquisition of additional driver mutations, such as FLT3 and NRAS. Clinical evidence shows that sAML patients exhibit resistance to conventional chemotherapies and their prognosis is very poor. Treatment options for sAML patients include intensive chemotherapy, targeted therapies, or 5-Aza/Venetoclax (BCL2 inhibitor). However, those sAML patients tend to be older and have critical comorbidities, and therapeutic options are limited. Therefore, the development of novel therapeutic approaches to overcome these critical issues in therapies for sAML is urgently needed to improve the outcome and to prevent the disease progression of MDS to sAML. In our previous reports, we showed that: (1) MDS-related mutations induce metabolic rewiring in hematopoietic stem/progenitor cells (HSPCs) resulting in suppression of mitochondrial TCA cycle and OXPHOS and activation of glycolysis, and (2) this metabolic rewiring causes hypermethylation of epigenomes and activation of HIF1A through inhibition of α-ketoglutarate (α-KG)-dependent dioxygenases and clonal advantage of MDS clones and plays a central role in MDS pathophysiology.
Notably, there are strong evidence showing that AML cells are highly relying on mitochondrial activities and actively utilize more fatty acids and glutamine in fatty acid oxidation (FAO) and glutaminolysis (GL) pathways, which may reflect their active metabolic state compared to that of MDS. Thus, we hypothesize that sAML has a distinct metabolic state compared to MDS and the metabolic reprogramming by FLT3 and NRAS signaling leads to the transformation from MDS to sAML, which in turn provide a therapeutic vulnerability in sAML. We therefore proposed to determine how FLT3 and NRAS signaling induce metabolic rewiring in our novel MDS and sAML model mice by performing stable isotope-labeling NMR and mass spectrometry analysis, and to elucidate transcriptional networks and signaling pathways which playing a critical role in metabolic reprogramming in sAML by integrating data of transcriptome analysis to metabolomics data. Furthermore, based on the data of metabolomics and transcriptome analysis, we proposed to test the efficacy of metabolic intervention by available inhibitors in MDS/sAML chimeric bone marrow transplantation mice, a model for transformation process of MDS to sAML. We are also going to test the metabolic interventions in HSPCs of high-risk MDS and sAML patients in vitro and in vivo so to provide potential preclinical data for a proposal of future clinical studies.
We have performed a series of in vitro and in vivo experiments in which we have found that: 1) Low TCA-cycle components in half of MDS patients group which are TGF-beta “active” and in mouse HSPCs harboring an MDS-related MLL gene mutation; 2) A novel mouse model knocking- down of a TCA-cycle enzyme SDH (succinate dehydrogenase, a.k.a. mitochondria complex II) develops full spectrum of MDS phenotypes which strongly suggests that MDS is a condition with metabolic dysregulation; 3) KD of SDH significantly reduces complex II and super complexes of OXPHOS; 4) Mouse and human sAML cells are sensitive to the inhibition of FAO and OXPHOS pathways; 5) Mouse sAML cells have elevated CPT1A levels; 6) CPT1A expression, but not CPT2 expression, has a trend of a worse outcome in TCGA dataset for AML; 7) We have been developing an unperturbed stable isotope-labeling NMR and Mass/Spec analysis, which will be used to determine how FLT3 and NRAS signaling induce metabolic rewiring in our MDS and sAML models in collaboration with Dr. Eric Pietras at U of Colorado.
In sum, we have made significant progresses on the aims that we proposed despite facing the pandemic and relocation. By completing all proposed studies in year 3, we expect to elucidate the detailed mechanisms of metabolic reprogramming in the progression of MDS to sAML and to develop rationale- based novel therapeutic strategy for sAML patients as well as high-risk MDS patients to prevent their transformation to sAML and/or eliminate sAML clones, which may also benefit other types AML patients.